Genetic Analysis and Viral Evolution of SARS-CoV-2: Impacts and Spread
"Evolutionary Phylogeny of SARS-CoV-2 and Its Impact on Public Health"
"INTRODUCTION
Over the past 2 decades, coronaviruses (CoVs) have been associated with significant disease outbreaks in East Asia and the Middle East. The severe acute respiratory syndrome (SARS) and the Middle East respiratory syndrome (MERS) began to emerge in 2002 and 2012, respectively. Recently, a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), causing coronavirus disease 2019 (COVID-19), emerged in late 2019, and it has posed a global health threat, causing an ongoing pandemic in many countries and territories (1).
Health workers worldwide are currently making efforts to control further disease outbreaks caused by the novel CoV (originally named 2019-nCoV), which was first identified in Wuhan City, Hubei Province, China, on 12 December 2019. On 11 February 2020, the World Health Organization (WHO) announced the official designation for the current CoV-associated disease to be COVID-19, caused by SARS-CoV-2. The primary cluster of patients was found to be connected with the Huanan South China Seafood Market in Wuhan (2). CoVs belong to the family Coronaviridae (subfamily Coronavirinae), the members of which infect a broad range of hosts.
The COVID-19 pandemic does not have any novel factors, other than the genetically unique pathogen and a furthe possible reservoir. The cause and the likely future outcome are just repetitions of our previous interactions with fatal coronaviruses. The only difference is the time of occurrence and the genetic distinctness of the pathogen involved. Mutations on the RBD of CoVs facilitated their capability of infecting newer hosts, thereby expanding their reach to all corners of the world (85). This is a potential threat to the health of both animals and humans. Advanced studies using Bayesian phylogeographic reconstruction identified the most probable origin of SARS-CoV-2 as the bat SARS-like coronavirus, circulating in the Rhinolophus bat family (86).
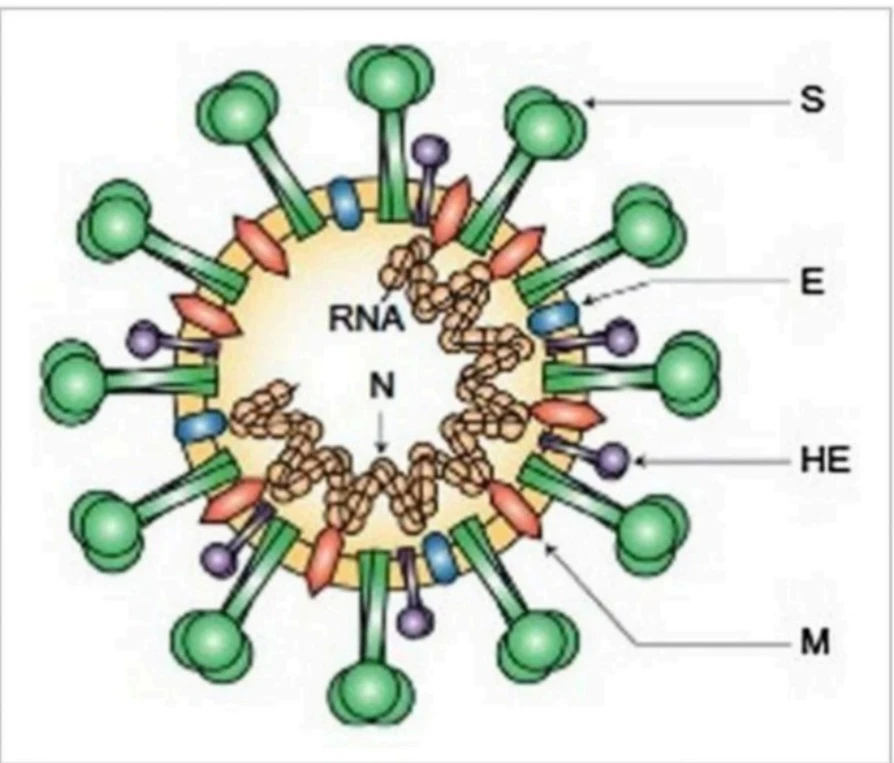
Phylogenetic analysis of 10 whole-genome sequences of SARS-CoV-2 showed that they are related to two CoVs of bat origin, namely, bat-SL-CoVZC45 and bat-SL-CoVZXC21, which were reported during 2018 in China (17). It was reported that SARS-CoV-2 had been confirmed to use ACE2 as an entry receptor while exhibiting an RBD similar.
Increasing reports of SARS-CoV-2 in sewage and wastewater warrants the need for further investigation due to the possibility of fecal-oral transmission. SARS-CoV-2 present in environmental compartments such as soil and water will finally end up in the wastewater and sewage sludge of treatment plants (328). Therefore, we have to reevaluate the current wastewater and sewage sludge treatment procedures and introduce advanced techniques that are specific and effective against SARS-CoV-2.
Since there is active shedding of SARS-CoV-2 in the stool, the prevalence of infections in a large population can be studied using wastewater-based epidemiology.
Recently, reverse transcription-quantitative PCR (RT-qPCR) was used to enumerate the copies of SARS-CoV-2 RNA concentrated from wastewater collected from a wastewater treatment plant (327). The calculated viral RNA copy numbers determine the number of infected individuals.
primary anti-genic epitopes mainly those recognised by neutralising antibodies.
The spike S-protein being in a spike form is subjected to a structural rearrangement process so that fusing the outer membrane of the virus with the host-cell membrane becomes easier. Recent SARS-CoV work has also shown that the membrane exopeptidase ACE enzyme (angiotensin-converting enzyme) functions as a COVID-19 receptor to enter the human cell.